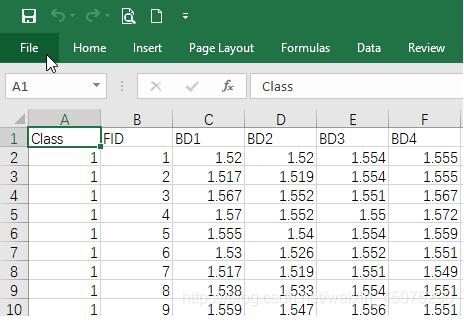
批量地做就是解放双手地过程,本文主要介绍了R语言批量读取某路径下文件内容的方法,文中通过示例代码介绍的非常详细,对大家的学习或者工作具有一定的参考学习价值,需要的朋友们下面随着小编来一起学习学习吧
R刚入门的时候,能够正确读取单个文件就觉得小有成就,随着时间的积累,单一文件地读取已经不能满足需求了,此时,批量地做就是解放双手地过程。
使用for循环把下载地TCGA数据读入R语言并转换成数据框
使用三个for循环来完成,这是第一个for循环。
1. 把所有数据读入在一个文件夹中
dir.create("data_in_one") #创建目标文件夹,也可右键创建
dir("rawdata/") #查看原路径的内容
for (dirname in dir("rawdata/")){
## 1.要查看的单个文件夹的绝对路径
mydir <- paste0(getwd(),"/rawdata/",dirname)
## 2.找到对应文件夹中的文件并提取名称,pattern表示模式,可以是正则表达式
file <- list.files(mydir,pattern = "*.counts")
## 3.当前文件的绝对路径是
myfile <- paste0(mydir,"/",file)
## 4.复制这个文件到目的文件夹
file.copy(myfile,"data_in_one")
}2. 寻找TCGA ID并让文件名称和TCGA ID保持一致。
第二个for循环。文件名称和TCGA ID的对应关系,藏在了metadata中。
metadata <- jsonlite::fromJSON("data/metadata.cart.2021-05-28.json")
metadata_id <- metadata[,c("file_name","associated_entities")]
## 1.准备容器,已经存在,我们把新数据添加在第三列
metadata_id
## 2.循环操作
for (i in 1:nrow(metadata_id)){
print(i)
metadata_id[i,3] <- metadata_id$associated_entities[i][[1]]$entity_submitter_id
}
## 重新命名
colnames(metadata_id)[3] <- "TCGA_id"
行排序,为了把文件名称和TCGA_id对应起来。读入的顺序和复制到新路径的顺序不一致,这一步的目的是让其保持一致。
rownames(metadata_id) <- metadata_id[,1]
metadata_id <- metadata_id[files,]
3. 输入文件名并提取文件的第二列(counts列)
#install.packages("data.table")
#构建函数
myfread <- function(files){
data.table::fread(paste0("data_in_one/",files))$V2
}
## 测试文件
test <- myfread(files[1])
4.1 使用for循环来批量读入并整合到一个数据框。
## 1.创建容器
gene_id <- data.table::fread(paste0("data_in_one/",files[1]))$V1
expr_df <- data.frame(gene_id=gene_id)
## 2.按照列读入
for (i in 1:length(files)){
print(i)
expr_df[,i+1] = myfread(files[i])
}
## 增加列名
colnames(expr_df) <- c("gene_id",metadata_id$TCGA_id)
### 意外发现
tail(expr_df$gene_id,10)
### 去掉最后5行
(nrow(expr_df)-5)
expr_df <- expr_df[1:(nrow(expr_df)-5),]
save(expr_df,file = "output/BRCA_RNASEQ_exprdf.Rdata")4.2 使用lapply + function 模式
1.函数
myfread <- function(files){
data.table::fread(paste0("data_in_one/",files))$V2
}
### 2.lapply
dd = lapply(files,myfread)
### 3.do.call
expr_df = as.data.frame(do.call(cbind,dd))
### 4.添加名称
colnames(expr_df) = metadata_id$TCGA_id
rownames(expr_df) = data.table::fread(paste0("data_in_one/",files[1]))$V1到此这篇关于R语言批量读取某路径下文件内容的方法的文章就介绍到这了,更多相关R语言批量读取文件内容请搜索编程学习网以前的文章希望大家以后多多支持编程学习网!
沃梦达教程
本文标题为:R语言批量读取某路径下文件内容的方法


猜你喜欢
- Go Web开发进阶实战(gin框架) 2023-09-06
- R语言关于二项分布知识点总结 2022-11-30
- Golang http.Client设置超时 2023-09-05
- R语言绘图数据可视化pie chart饼图 2022-12-10
- Ruby on Rails在Ping ++ 平台实现支付 2023-07-22
- 汇编语言程序设计之根据输入改变屏幕颜色的代码 2023-07-06
- Swift超详细讲解指针 2023-07-08
- R语言-如何切换科学计数法和更换小数点位数 2022-11-23
- Ruby 迭代器知识汇总 2023-07-23
- Ruby的字符串与数组求最大值的相关问题讨论 2023-07-22